


L'IVDR n'est pas pour nous, nous sommes accrédités par le COFRAC !
Confusion entre la législation européenne et l'accréditation/Certification Professionnelle : un dilemme aux lourdes conséquences pour les laboratoires ayant des procédés de DIV internes
Le règlement européen 2017/746 sur les dispositifs médicaux de diagnostic in vitro (IVDR) a rapidement été perçu comme un obstacle au développement et à l'innovation dans le secteur de la production des dispositifs médicaux.
Face à ce raz-de-marée réglementaire, les fabricants ont su réagir rapidement en formant des groupes de réflexion, organisant des formations, renforçant leurs équipes et luttant pour obtenir un report de ces nouvelles exigences.
À l'inverse, bien que soumis à l'article 5.5 de l'IVDR, les organisations de santé européennes n'ont pas bénéficié de ce même élan collectif pour sensibiliser, préparer et faciliter l'implémentation de cette réglementation dans les laboratoires concernés.
Les laboratoires n'ont été que peu impliqués dans le développement de l'IVDR, et rarement consultés.
En mai 2024, la FDA (Food and Drug Administration) américaine a publié une directive au but similaire, illustrant la volonté internationale de renforcer les réglementations sur les diagnostics in vitro internes.
Selon les articles 96 et 97 de l'IVDR, il est ainsi prévu que les États membres désignent des autorités compétentes pour superviser et contrôler ces exigences.
En France, la nomination de l'autorité compétente est en cours au ministère de la Santé. Il semblerait possible que les Agences Régionales de Santé (ARS) se voient confier cette responsabilité.
Dans d'autres pays européens comme l'Allemagne, l'Autriche ou l'Italie, les mêmes autorités que celles responsables de la vigilance des produits de DIV et de leurs fabricants ont été choisies.
Certaines sociétés savantes, comme la Société Européenne de Génétique, se sont penchées sur la question et ont informé leurs membres.
Néanmoins, il subsiste, en particulier dans le paysage francophone des laboratoires, une croyance erronée, presque comparable à une ‘fake news’. Cette croyance suggère qu'une accréditation professionnelle selon l'ISO 15189, une accréditation CLIA ou une accréditation par le COFRAC exempterait les laboratoires ayant des procédés de DIV internes de toutes autres exigences de l'IVDR.
Aperçu des Exigences de l'Article 5.5) de l'IVDR
Pour mieux aborder ce sujet, un aperçu des exigences de l'article 5.5 de l'IVDR permettra de démystifier ces demandes.
Que sont les DIV internes ?
Il existe plusieurs scénarios selon lesquels une procédure d'analyse diagnostique peut devenir un DIV interne. Ces scénari peuvent être divisés en quatre groupes :
1. Utilisation d’un produit marqué CE IVD en dehors de sa destination, de son utilisation ou des informations prévues par le fabricant : Lorsque le produit est utilisé pour des fins autres que celles spécifiées par le fabricant.
2. Utilisation d’un produit non marqué CE IVD (les RUO ou GLU) : Les produits destinés à la recherche uniquement (RUO) utilisés à des fins diagnostiques.
3. Combinaison de produits marqués CE IVD et non marqués : L’utilisation combinée de dispositifs marqués CE et de dispositifs non marqués dans une même procédure diagnostique.
4. Développement propre du laboratoire : Lorsque le laboratoire développe son propre produit et procédé diagnostique.

Les scénarios du DIV (source Platomics)
Ces scénarios rendent plus que plausible la possibilité pour les laboratoires de passer dans la catégorie des fabricants de DIV internes et d’être donc soumis à l’article 5.5) de l’IVDR.
En outre, avec les fabricants de DIV qui repensent actuellement leur gamme de produits, des changements de destination pour des DIV usuels sont à prévoir.
Une analyse des écarts (GAP analysis) est donc fortement recommandée pour les laboratoires, afin de réévaluer les paramètres de diagnostic qu'ils proposent à la lumière des exigences de l'IVDR.
Quelles sont les exigences de l’IVDR pour les DIV internes ?
Les laboratoires ayant donc reconnu l’applicabilité de l’article 5.5) de l’IVDR se trouvent alors confrontés aux exigences réglementaires de cet article, s’ils souhaitent continuer à utiliser des DIV internes dans leurs procédures de diagnostic.
À la première lecture, l’article 5 5) de l’IVDR parait tout simple et donne une impression primaire que les DIV internes sont presque exempts de la plupart des exigences de l’IVDR, sauf si on lit en détail l’annexe I qui est applicable aux laboratoires depuis déjà mai 2022. Et l’annexe I renvoie à toutes les obligations de fabricants de DIV en général.
En effet, l’article présente 9 exigences qui devraient déjà être mises en place par les laboratoires, selon le calendrier du règlement (dès mai 2022 jusqu'à 2030 pour le point d) ) :
• a) Pas de cession des produits à une autre entité légale distincte : les autorités nationales compétentes sont responsables, si besoin est, de clarifier la notion nationale d’entité légale.
• b et c) Un système de management de la qualité conforme à l’EN ISO 15189 et, si applicable, les exigences nationales concernant l’accréditation et la certification des laboratoires, ainsi qu'un système de management de la qualité couvrant le développement et la production des DIV internes concernés (y compris la traçabilité, la vigilance et surveillance des produits concernés). L’article 10 de l’IVDR qui peut être utilisé comme une recommandation pour ces activités, décrit les aspects minimaux qu’un tel système doit couvrir, en particulier au regard des risques potentiels (pour l’utilisateur, pas juste pour le patient)
• d) L’organisation de santé justifie dans sa documentation que les besoins spécifiques du groupe cible de patients ne peuvent pas, ou ne peuvent pas à un niveau équivalent, être satisfaits par un produit sur le marché des dispositifs médicaux.
• e) L’organisation de santé doit mettre à disposition des autorités compétente, à leur demande, des informations sur l’utilisation des produits concernés. Cette information doit aussi contenir une justification sur la production, les changements et l’utilisation des produits.
• f) Une déclaration rendue publique et contenant des points obligatoires doit être faite par l’organisation de santé (y compris la mention que les produits remplissent les exigences de l’annexe I du règlement IVDR).
• g) Le site de production, les procédés de fabrication, les données de performances des dispositifs internes correspondant à la classe D des risques ainsi que leur destination doivent être présentés de manière détaillée, afin que les autorités compétentes puissent s’assurer de la conformité à l’annexe I. Ce point peut être étendu par les Etats membres aux dispositifs de classe A, B et C.
• h) Toutes les mesures nécessaires doivent être prises par l’organisation de santé pour assurer que la production de tous les produits concernés soit faite selon la documentation demandée au point ci-dessus.
• i) Un système de mesures correctives et de surveillance des produits concernés doit être mis en place. L’organisation doit évaluer les expériences venant de l’utilisation des DIV internes
Ces différentes exigences correspondent à des échéances pour les prochaines années.
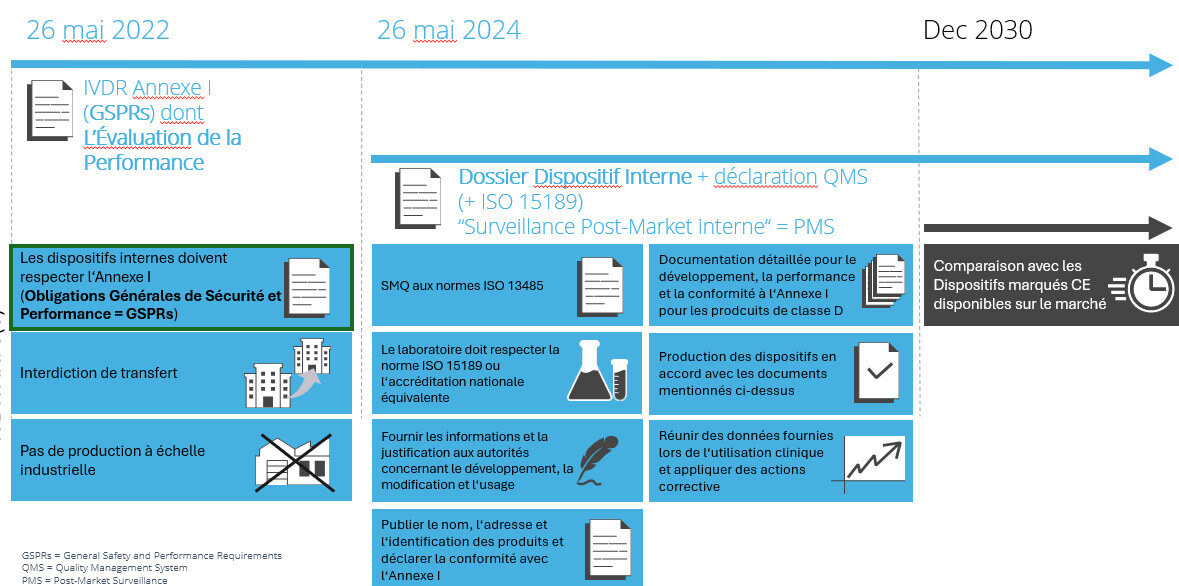
Exigences et échéances copyright Platomics GmbH
De plus, en janvier 2023, un MDCG (Medical device coordination group) document (MDCG 2023-1) a enfin été publié pour éclaircir certains points laissant le doute dans l’article 5. 5). Bien sûr, ces documents du groupe de coordination des dispositifs médicaux n’ont pas de force légale obligatoire, mais dans le contexte européen du règlement européen IVDR (Article 99), il n’est pas pensable de les ignorer.
La particularité de l’Annexe I
Pour tous les types de DIV et de fabricants, l'Annexe I est la porte d'entrée des exigences de la documentation technique et, en même temps, une carte de visite pour les organismes notifiés et les autorités compétentes. En effet, cette annexe peut être vue comme une liste de vérification ou un index pour les informations documentant les exigences de l'IVDR.
Cette annexe regroupe plus de 190 points que les fabricants peuvent avoir à satisfaire, quels que soient leurs produits- DIV internes ou marqués CE.
À la lecture de cette annexe, il est clair que ces nombreux points sont orientés vers les fabricants de produits DIV marqués CE, qui développent, produisent et distribuent leurs produits sur le marché.
Cependant, pour les DIV internes, de nombreuses exigences placent les organisations de santé devant des demandes matériellement très difficiles à réaliser (par exemple, les exigences relatives au "software life cycle" ou à la sécurité électrique des appareils).
La liste de vérification de l’annexe I renvoie néanmoins à des documents prouvant la conformité à ces exigences qui doivent être fournis et préparés par les organisations de santé. L'absence de ces documents entraînerait une non-conformité à l'IVDR, rendant l'utilisation des DIV internes illégale pour des activités de diagnostic.
Le positionnement de l’IVDR face aux accréditations professionnelles
En Europe, bien que l'IVDR soit un règlement juridiquement directement applicable en droit national sans nécessité de transposition légale, il persiste des différences notables selon les pays et les disciplines de biologie médicale.
Lors de pré-audits préparatoires à l'accréditation et dans le cadre d'une analyse de l'environnement réglementaire, les laboratoires ou organisations de santé avancent souvent l'argument que leurs procédés, procédures, paramètres de diagnostic ou systèmes de qualité, agréés ou accrédités par des organisations nationales et internationales (par exemple, le Cofrac ou le College of American Pathologists selon l'ISO 15189 ou le CAP Laboratory Accreditation Program), confèrent automatiquement une conformité à l'IVDR.
Cette hypothèse est néanmoins incorrecte. Bien que ces accréditations par des organisations internationales et nationales soient des gages d’excellence de la qualité et du professionnalisme pour les organisations de santé concernées, elles ne répondent pas aux exigences du droit européen applicable sous l'IVDR.
Un système de qualité conforme à l'ISO 15189 est certes une des conditions de l'article 5.5 de l'IVDR, mais il ne suffit pas à lui seul comme le souligne le MDCG 2023-1, page 12 Point 3.5.2: “compliance with EN ISO 15189 alone does not constitute an appropriate QMS for the manufacture of in-house IVDs.” De nombreuses autres conditions et exigences, non couvertes par ce standard ni par aucun autre système d'accréditation des organisations de santé, doivent être remplies.
Le lien étroit des fabricants de DIV internes et l’EN ISO 13485 :2016
Un dernier point qui surprend souvent dans le monde du diagnostic et du laboratoire : l’IVDR, en soumettant les organisations de santé à des exigences de contrôle de la production, les renvoie nécessairement à un système de qualité couvrant selon le point b de l’article 5 5) la production ainsi que les risques inhérents à l’utilisation du DIV interne (selon l’ISO 22367:2020 pour le management du risque dans les laboratoires médicaux ou l’ISO 14971:2019 pour la gestion des risques pour les fabricants de DIV).
Dans ce domaine l’EN ISO 13485:2016 est le seul standard couvrant ces procédures pour les fabricants de DIV. Il parait donc inévitable pour les organisations de santé de s’y référer.
En Résumé
L'IVDR introduit des changements significatifs qui affectent les laboratoires de diagnostic in vitro. Il est essentiel de comprendre les nouvelles exigences, de préparer une stratégie de mise en conformité et de rester informé des développements internationaux en matière de réglementation.
| Ce que vous devez savoir : • Les exigences de l’IVDR ne sont pas connectées aux exigences d’une certification professionnelle. • Les laboratoires doivent déjà être à ce jour conformes à l'annexe I de l'IVDR et remplir les GSPR (General Safety and Performance Requirements). • Un système de qualité selon l’ISO 15189 doit être mis en place dans les laboratoires (sans obligation d’accréditation), mais une accréditation ne suffit pas pour être conforme à l'article 5.5 de l'IVDR. • Les procédés de production et de développement doivent idéalement s’aligner sur les normes ISO 13485, car ces procédés ne sont pas couverts par l' ISO 15189 tout comme l’implémentation et l’intégration de procédure pour couvrir toutes les exigences de l’Article 5.5). Par exemple : la révision des expériences issues de l’utilisation clinique des DIV internes. (voir page 11 MDCG 2023-1 “ the health institution shall review experience gained from clinical use of the device and take all necessary corrective actions “ • L’analyse des risques doit être réalisée au niveau du produit, de l’utilisateur et de son environnement, pas juste en relation avec le patient. |
Il est donc urgent pour les laboratoires d'agir et de trouver des solutions viables, afin de ne pas compromettre la disponibilité des tests diagnostiques sur le marché, ce qui pourrait mettre en danger la santé publique, particulièrement dans les disciplines jeunes et innovantes du diagnostic moderne.
A côté des systèmes de management de la qualité usuels basés sur une documentation papier et beaucoup de tableaux Excel, il existe sur le marché européen des solutions digitales qui incluent les avancées de l’intelligence artificielle et qui peuvent être envisagées par les organisations de santé, afin d’utiliser au mieux les ressources de personnel dans des tâches non-cliniques.
Il est en effet critique de trouver des solutions pour les laboratoires rencontrant déjà des difficultés de ressources de personnel et qui ne peuvent pas toujours faire face aux nouvelles demandes règlementaires au niveau européen.
Auteur :
Marie Salin, Auditrice- Consultante
Certifiée TÜV auditor for medical devices, ISO 13485 ISO 15189
20 ans d’expérience dans le monde du DIV (West Medica et Platomics)
marie.salin@platomics.com
Références:
MDCG 2023-1 - Guidance on the health institution exception under Article 5(5) of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 - January 2023 - European Commission (europa.eu)
Ordonnance n° 2022-1086 du 29 juillet 2022 portant sur l'adaptation du droit français au règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro
ISO 22367:2020 Laboratoires de biologie médicale - Application de la gestion des risques aux laboratoires de biologie médicale
Partager cet article : |


